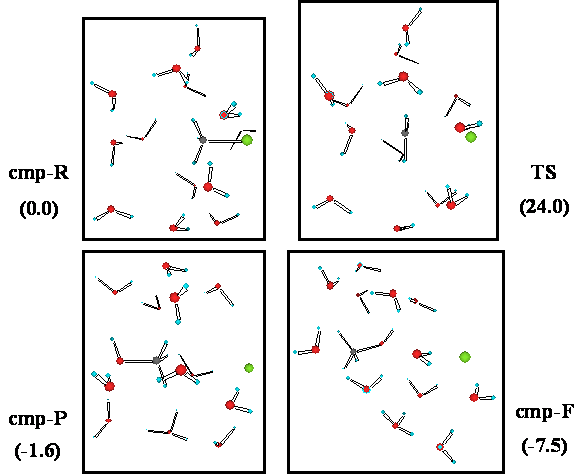
図1.水13個を含む系の塩化メチルの加水分解反応の始状態(cmp-R),遷移状態(TS),終状態(cmp-P),最安定構造(cmp-F)。括弧内は相対エネルギー(kcal/mol)。
溶液中において進行する反応のメカニズム
【1】塩化メチルの加水分解(ab initio MO計算)
塩化メチルのまわりに水分子を13個まで含めた系にたいして非経験的分子軌道法(ab initio MO法)計算を実行した。水の数をいろいろ変えて計算した結果,少なくとも3個の水分子を含むことが,SN2型置換反応の反応形式の特徴をあらわすためには必要であることがわかった。また,反応エネルギーや同位体効果の計算値は,まわりに13個の水分子をあらわに含んだ系で,実測値をよく再現できることがわかった。この系の定常状態(反応の始状態,遷移状態,終状態,最安定構造)の構造と相対エネルギーを図1に示す。使用した計算レベルはHF/6-31+G*である。反応の過程のどの状態においても,反応する水分子と脱離したCl−イオンの間に溶媒水分子を介した水素結合ネットワークが形成されていることがわかる。13個の水分子を含んだ系ではさまざまな実測値を再現でき,水分子数がそれより少ない系では再現できない,ということは,遷移状態において13個の水分子が反応中心を取り囲むようにして存在することが反応の進行に本質的に関わっていることを示している [1]。
図1.水13個を含む系の塩化メチルの加水分解反応の始状態(cmp-R),遷移状態(TS),終状態(cmp-P),最安定構造(cmp-F)。括弧内は相対エネルギー(kcal/mol)。
塩化メチルの加水分解反応に対して,水の数をいろいろ変えて計算した結果,3個の水分子を含む系では,反応熱や同位体効果は実測値にはあわないが,水素結合のネットワークやプロトン移動の様子は,SN2型置換反応の特徴をあらわしていることがわかった。この系における反応のIRC(Intrinsic Reaction Coordinates)にそったエネルギー変化や原子間距離のプロットを図2に示す。IRCにそったプロットには,攻撃してきた水分子から隣の溶媒水分子のプロトン移動は遷移状態をすぎた後におこり,その次の水分子へのプロトン移動は段階的におこることが表れている。しかし,IRCの解析では時間的なことはわからない。加水分解における段階的な反応がどのような時間経過で進行するのかに関する情報は,次の節で述べるような他の手法を用いることによって初めて得ることができる。
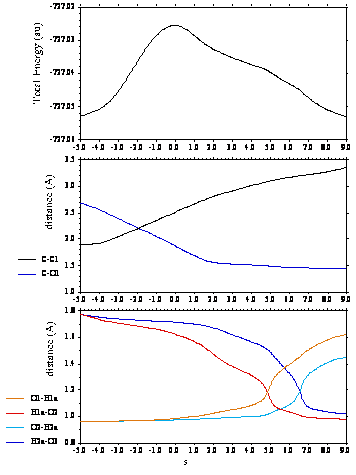
図2.水分子が3個の場合のCH3Clの加水分解反応における,IRCにそったエネルギーや水素結合距離の変化。O1:炭素原子に攻撃する水分子の酸素原子,O2:その隣の水分子の酸素原子,O3:さらに隣の水分子の酸素原子,H1a:O2にプロトンとして移動していく水素原子,H2a:O3にプロトンとして移動していく,元はO2に結合している水素原子。
【2】塩化メチルの加水分解(direct ab initio MD計算)
加水分解の際に,攻撃する水分子から溶媒水分子に,どの段階でプロトン移動がおこるのであろうか。これを明らかにするためには,反応の時間変化を見ることが必要である。そのためには分子動力学(MD)法を用いることがふさわしい。しかし通常のポテンシャル関数を用いたMD法は反応過程を明らかにするために使うことはできない。ポテンシャル関数は反応をあらわすために使うことができるほど信頼性がないからである。そこで,反応の1ステップごとにab initio MO計算をおこなう direct ab initio MD法を実行した [2]。
3個の水を含む系での塩化メチルの加水分解反応が時間とともにどのように進行するのかを見るために,反応の遷移状態からdirect ab initio MD法計算をスタートさせた(図3)。direct ab initio MD法計算は,MD計算の各ステップにおいて,ab initio MO計算によってエネルギーおよびその微分の計算を実行するものであり,いわば,Born-Oppenheimer近似に基づいたシミュレーションである。ある瞬間において,そのときの原子核の配置にしたがって電子の状態が定まる。その次の瞬間においては,原子核の動きに電子は瞬時に応じて,そのときの原子核の配置にしたがって電子の状態が定まる。これが繰り返されていく。原子の動きを古典的にシミュレートし,その原子核の配置に応じた電子の状態やエネルギーを ab initio MO計算で求める,という手法が,direct ab initio MD法である。
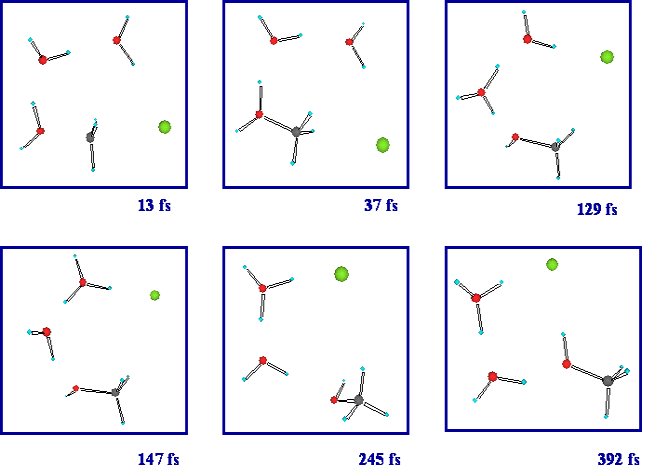
図3.塩化メチルの加水分解反応のスナップショット
ステップごとにHF/6-31Gレベルのab initio MO法計算を実行するdirect ab initio MD法計算の結果,水分子が中央の炭素原子の背後から近づいた後,ほぼ遷移状態に近い構造で数十fsecの間とどまり,その後,プロトンが移動していく様子が見出された。塩化メチルの加水分解反応の場合,攻撃する水分子から置換されて出て行く塩素原子まで水素結合のネットワークが形成されており,その水素結合のネットワークにそってプロトンが移動していく。水分子が3個の系が,その様子を解析することのできる最小の系である。
原著論文:
[1] Hiroshi Yamataka and Misako Aida, "An ab initio MO study on the hydrolysis of methyl chloride," Journal of Molecular Structure (Theochem), 461-462, 417-427 (1999).
[2] Misako Aida, Hiroshi Yamataka and Michel Dupuis, "Ab initio MD simulations of a prototype of methyl chloride hydrolysis with explicit consideration of three water molecules: a comparison of MD trajectories with the IRC path," Theoretical Chemistry Accounts, 102, 262-271 (1999).
[3]Masato Tanaka and Misako Aida, "An Ab Initio MO Study on Orbital Interaction and Charge Distribution in Alkali Metal Aqueous Solution: Li+, Na+ and K+," Journal of Solution Chemistry, in press (2004).
![]()