direct ab initio MDからの振動スペクトル
非経験的分子軌道法の汎用的なプログラムでは、分子の構造最適化や基準振動解析がルーチン的に計算できるようになっている。計算で得られた分子の構造は、使用する基底関数や計算手法のレベルを選べば、実測値をかなりよく再現することが知られている。振動数については、非調和項を得るための試みもなされているが、非経験的分子軌道法による振動解析の多くは、基準振動解析である。非調和項は小さいものとして無視されることが多い。
分子動力学(MD)法は、物理学・化学・生物学の分野において、構造など系の性質の時間変化やゆらぎを計算できる手法として、最近多く使用されるようになっている。分子に対する分子動力学法によるトラジェクトリーから得られる振動数は、非調和項を含むものとして解釈される。しかし、どの程度非調和項を含んでいるのか、実際には意識されないことが多いと思われる。
本研究は、ab initio MD法の結果得られる振動数から、どのようにして正確な非調和項を得ることができるのかを明らかにすることを目的としている。基準振動解析からは得られない描像を得ることができることを示す。IRとRaman スペクトルの振動数および強度を,direct ab initio MD法に基づいて計算する。基音だけでなく結合音や倍音も求める。
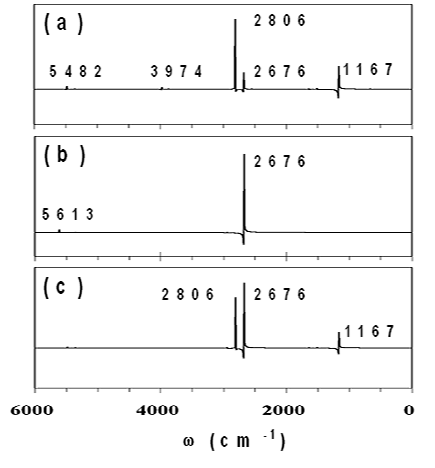
Direct ab initio MD計算の各ステップにおいて双極子モーメントと分極率テンソルを計算し、その時間変化の自己相関関数のフーリエ変換をすることによって、IRとRamanスペクトルの振動数だけでなく強度を求めることができることを示した。図は、D2O分子に対して,MP2/aug-cc-pVDZの計算レベルでの非経験的分子軌道法計算に基づくDirect ab initio MD計算をすることによって得られた、IRスペクトル(a)、Ramanスペクトル(b) およびdensity of states (c) である。
原著論文:
[1] Misako Aida and Michel Dupuis,"IR and Raman intensities in vibrational spectra from direct ab initio molecular dynamics: D2O as an Illustration," Journal of Molecular Structure (Theochem), 633, 247-255 (2003).
[2] Misako Aida and Michel Dupuis, "Fundamental absorption frequency from quasi-classical direct ab initio molecular dynamics: Diatomic Molecule," Chemical Physics Letters, 401, 170-174 (2005).
![]()